研究テーマ
・「細胞競合」の視点から、がんや病気の起こりのメカニズムを理解し、予防技術実現につなげる
・生物多様性データと超短命魚を利用して、ヒトの老化・寿命の予測・制御を狙う
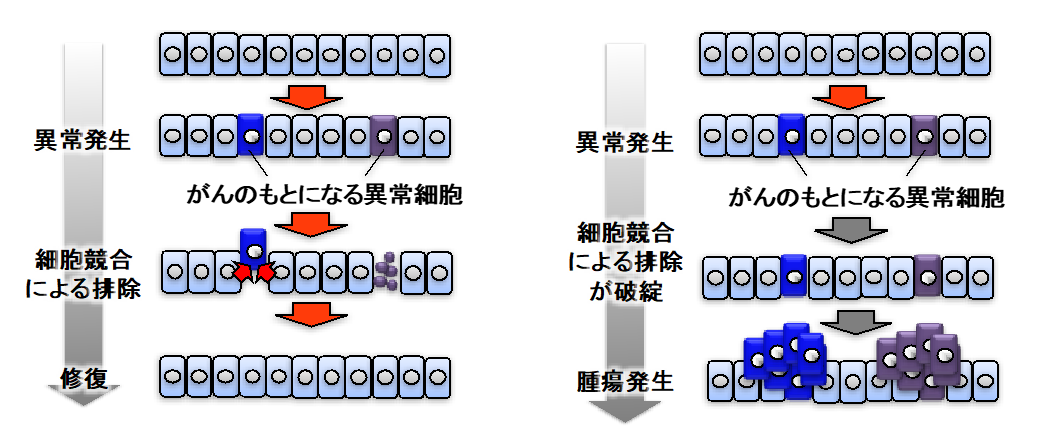
ヒトを含む多細胞生物は常に撹乱(ノイズ)にさらされています。例えば、細胞分裂の過程で複製エラーを起こしますし、代謝によって生じた活性酸素などがゲノムを傷つけることもあります。多細胞生物はこうした不可避なノイズに常に晒されており、それを何らかの方法で克服することで、正確な発生・再生や恒常性維持を成し遂げているはずです。私たちは最近、「細胞競合」と呼ばれる現象がこのノイズ除去を担っていることを発見しました(Nature Communications 2019, 2022, 2024; Science Advances 2024)。細胞競合は、細胞集団内に生じたエラー細胞(病的な細胞)を細胞間コミュニケーションによって感知し、排除する仕組みです。このメカニズムが破綻すると、病的な細胞が蓄積し、不適切な場所に細胞が配置されたり(脊髄の細胞が脳にできたり)、運動機能がうまく発達しなかったり、がんが生じたり、短寿命になったりします。私たちは現在、この細胞競合のメカニズムを解明するとともに、ヒト疾患・がんの発症との関連を解析しています。そしてこの知見を利用して、病気やがんの予防技術に繋げていきたいです。
また、老化・寿命の予測・制御も目指しています。老化を抑制できれば、あらゆる加齢性疾患の発症を抑制できるはずです。これまでの老化研究では、データをとるのに時間がかかる(ヒトの老化過程を解析すると何十年もかかり、マウスであっても3年かかる)ことがその進捗を阻んできました。私たちは最近、超短命魚キリフィッシュ(寿命3〜6ヶ月)を利用することで、短期間での老化解析を実現しました(Sci Rep 2022; NPJ Aging 2024; Scinece Advances 2024)。さらに、AMEDや民間財団のご支援のもと、100歳・110歳以上の長寿のヒトや、数百年生きるサメなどの生物多様性データから長寿因子を探り、それをキリフィッシュに導入して健康寿命延伸物質を見つけ出す研究を進めています。さらには、健康診断で残寿命や未来の老化速度を予測する技術の確立も目指しています。
最新の研究内容については以下をご覧ください(色文字をクリックいただくとリンク先に行けます)。
プレスリリース(モルフォゲン修復機構Nature Commun 2019; がん研究Nature Commun 2022; 老化研究Sci Rep 2022; 初期胚体軸誘導の進化Nature Commun 2023; 老化によるシグナル空間制御の破綻NPJ Aging 2024; 生殖細胞が寿命の性差を決める Science Advances 2024; 細胞社会の秩序は、フォースによって守られる Science Advances 2024; 細胞競合が臓器形成のしくじりを修正す Nature Commun 2024; 小胞体ストレスが皮膚幹細胞を若い状態に戻す Aging Cell 2025)
2019年以降メディア報道された成果については以下をご覧ください(一般向け。色文字をクリックいただくとリンク先に行けます)。
ナゾロジー 2025年10月 ストレスで「肌が若返る」可能性、超速老化魚の研究で判明
日刊工業新聞 2025年10月 小胞体ストレスで皮膚活性 阪大など、老化制御解明に前進
医療NEWS 2025年10月 網膜ジストロフィーの多発形態異常合併、CDK9活性と関連-成育医療センターほか
産経新聞 2025年3月 おなかに目ができないのはなぜか 「不良細胞」排除の仕組みを解明、がん治療への応用期待
佐賀新聞、山陰中央新報デジタル、山陽新聞、神戸新聞、新潟日報デジタル、静岡新聞デジタル、福井新聞、東奥日報、下野新聞デジタル、熊本日日新聞、沖縄タイムス、愛媛新聞オンライン、日本海新聞 2025年3月 がんの起源はどこか 見えてきた細胞集団の暴走、始まりの年齢
朝日新聞 朝刊(2/4) 2025年2月 ~がんとともに~ 異常細胞 増殖の過程に迫る
朝日新聞デジタル 2025年2月 がんの起源はどこか 見えてきた細胞集団の暴走、始まりの年齢
時事通信 2024年12月 「細胞競合」で不良除去 脊髄や筋肉の形成過程―大阪大
日本経済新聞オンライン 2024年12月 異常細胞除く「細胞競合」、因子特定で可視化 大阪大
日本経済新聞 2024年12月 異常細胞除く「細胞競合」
科学新聞 2024年12月 健康体維持に細胞間張力利用 不良細胞を感知・排除
日本経済新聞 2024年12月 細胞同士が異常を「監視」
日本経済新聞オンライン 2024年12月 細胞同士が「監視」し異常除去、がん予防応用も 大阪大
朝日新聞デジタル 2024年11月 引っ張る力で不良細胞を感知・排除する仕組み発見 大阪大
イギリス ガーディアン紙 2024年6月 Why do women outlive men? Cells that develop into sperm and eggs could give the answer
米国ニューズウィーク紙 2024年6月 Vitamin May Extend Lifespan, New Research Suggests
朝鮮日報 2024年6月 내시가 장수한 이유는 거세, 日과학자 동물실험으로 입증
日刊工業新聞 2024年6月 脊椎動物の生殖細胞、寿命の性差生み出す 阪大が特定
日経バイオテク 2024年6月 大阪大、オスよりメスが長命な仕組みを魚の実験で解明
朝日新聞デジタル+朝日新聞 夕刊(6/13) 2024年6月 メスはなぜ長生き? 寿命を精子は縮め、卵子は延ばす 阪大など実験
読売新聞オンライン+読売新聞 朝刊(6/16) 2024年6月 精子できなくすると一生が13%延びた…短命の魚で解明、雌雄の平均寿命差の理由の一つか
時事通信+時事メディカル 2024年6月 生殖細胞が寿命に影響=短命小魚で遺伝子操作実験―大阪大
産経新聞 2024年6月 長寿の秘訣は「卵子」にあり オスは精子で短命に…大阪大研究チームが魚で実験
読売新聞東京本社版 朝刊(6/24) 2024年6月 メス長寿 生殖細胞影響 大阪大など、魚で研究・発表 オスの寿命 精子できなくすると延び
産経新聞オピニオン 2024年6月 <浪速風>「生命の神秘」が解き明かされる日も期待したくなる研究成果
科学新聞 2024年6月 ビタミンD投与で小型魚の寿命延伸 阪大微研が発見
日本経済新聞 2024年6月 生殖細胞無くすと寿命変化 大阪大、魚類で確認
マイナビニュース 2024年6月 阪大、脊椎動物では卵子が寿命を延ばし精子が寿命を縮める可能性を発見
オーストラリア COSMOSマガジン 2024年6月 Are sperm and eggs responsible for the gender gap in life expectancy?
生殖細胞による寿命制御:海外メディアでも多数報道!!
EurekAlert! Asia Research News ScienMag Bioengineer.org Medical Xpress Technology Networks
ナゾロジー 2024年5月 3カ月で老化する!?不思議な魚が解き明かす老化の秘密
朝日新聞デジタル 2023年11月 ノックアウトではわからぬ遺伝子の謎 阪大チームが解明に使った技術
文藝春秋4月号 2023年3月 「老化は治療できるか」
週刊医学界新聞 2023年1月 老化研究を加速させる実験モデル生物ターコイズキリフィッシュ
日経産業新聞 2022年8月 老化の研究、魚で効率よく 大阪大、遺伝子改変を可能に
日経新聞電子版 2022年8月 老化の研究、魚で効率よく 大阪大学
日経バイオテク 2022年8月 大阪大、老化研究の新モデル“超速成長・超速老化魚ターコイズキリフィッシュ”の遺伝子機能高速解析系を開発
日経バイオテク 2022年3月 大阪大石谷教授、前がん細胞が排除されるか腫瘍を形成するかの分かれ道を解明
日経メディカルオンライン 2022年3月 前癌細胞が排除されるか腫瘍を形成するかの分かれ道は?
日経産業新聞 2019年11月 大阪大学、胚、異常避け育つ仕組み特定
日刊工業新聞 2019年10月 胚成長時の異常細胞排除、阪大など仕組み発見 先天性疾患治療に期待
以下が、現在を含め、これまで進めてきた研究テーマです。
現在進行中のテーマ
ヒトを含む動物の体内に生じた異常細胞が免疫システムによって排除され、これが健康な体の維持に貢献していることがよく知られています。
しかし近年、動物組織が免疫システムに頼らずに自らの力で異常細胞を排除する能力を有する(具体的には、上皮組織などにおいて異常な細胞が突発的に生じた際に、異常細胞の隣接正常細胞がこれを感知し、積極的に排除する)ことが明らかになりつつあります。
この現象は細胞競合と呼ばれ、がん発生の初期においてがんの発生を抑制する仕組みとして注目されています。
また、私たちは最近、この異常細胞排除システムが、動物の初期発生や器官形成などにおいても機能しており、正確な組織形態形成(発生プログラムのロバストネス)を支えていることも見出しています。
私たちはこの異常細胞排除システムに注目し、がんの初期発生メカニズムと、発生ロバストネスの研究を進めています。
Wnt/β-cateninシグナルは、動物種をこえて保存された(植物や単細胞生物にはない)シグナル伝達経路であり、前後軸(頭尾軸)・背腹軸などの体の極性形成やパターン形成、幹細胞・前駆細胞の運命制御などを担う「動物を動物たらしめるシグナルシステム」です。
近年では、細胞の代謝制御や老化にも関わっていることもわかってきています。
また、その制御破綻は、がんや糖尿病、肥満、骨粗鬆症、精神疾患など多様な疾患の発症に関わります。
私たちの研究室では、Wnt/β-cateninシグナルの未知の制御機構を探索することで組織の形態形成、恒常性維持のメカニズム解明に貢献するするとともに、その制御技術を開発して医療に貢献することも目指しています。
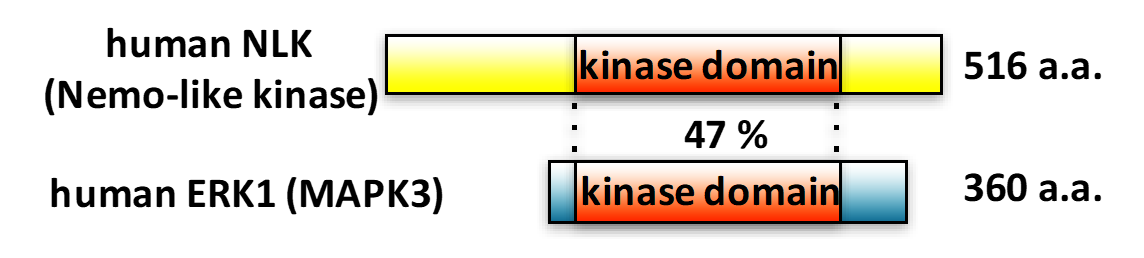
Nemo-like kinase (NLK)は動物種をこえて保存されたMAPKファミリーのSer/Thrキナーゼです。
MAPKファミリーに属するkinaseとしては、ERK、p38、JNKが有名ですが、NLKはこれのどれにも属さない特殊なkinaseです。
私たちはこれまでに、NLKがWntシグナルやNotchシグナルの制御を介して神経幹細胞・前駆細胞の運命を制御することを明らかにしています。
一方で近年、NLKが神経変性疾患やがんの発症に関わることが明らかになりつつあります。
私たちは、NLKが神経変性疾患やがんを促進するメカニズムを明らかにし、これを起点としてこれらの疾患の治療を行おうとしています。
 ヒトの病気のメカニズムを解明したり、治療薬を開発するためには、ヒトの病気を再現した実験動物(疾患モデル)を使用する必要があります。
ヒトの病気のメカニズムを解明したり、治療薬を開発するためには、ヒトの病気を再現した実験動物(疾患モデル)を使用する必要があります。
これまで、マウスやラットなどを疾患モデルとして用いた研究が精力的に行われてきましたが、これらの動物はヒト同様に哺乳類であるという利点がある一方で、解析にあたってのスループットや検体数に制限があり、また、生命倫理的な問題によっても研究の制限があります。
私たちは、ゼブラフィッシュなどの小型魚類を疾患モデルとして使った、多検体、ハイスループット、高解像度(イメージングとの併用)で、かつ、生命倫理問題を軽減した疾患研究を提案しており、国内外の医学薬学研究者と魚疾患モデルの開発を進めています。
特に、魚を使った「がんの発生過程のイメージング解析」に力を入れており、哺乳類を使った研究では明らかにすることができない深さ、精密さで“がん”を理解しようとしています。
過去の研究成果
私たちは、生化学的スクリーニングにより、NLKの新規基質としてNotch3を得た。Notch3はNotch1,2,3,4からなる膜貫通型受容体タンパク質Notchファミリーに属する分子である。Notchは隣り合う細胞に存在するリガンドよりシグナルを受け取ると、その細胞内領域を細胞質中へと切り離し、切り離された細胞内領域(Notch-ICD)は核内へ移行し、転写因子CSL (RBPJκ/Su(H))及び転写共役因子Mastermind (MAM)と三者複合体を形成し、標的遺伝子の転写を活性化する。また、このNotchシグナルは、幹細胞の維持や急性白血病発症に関与することが知られている。私たちはまず、NLKがNotch3をリン酸化してNotch3の活性に影響を与えるかを検討するために、Notch3-ICD(活性化型Notch3)による転写活性化に対するNLKの影響をレポーターアッセイにより調べた。残念なことに、HEK293あるいはヒト子宮頸癌由来細胞HeLa細胞においてNLKを過剰発現させても、Notch3-ICDによる転写活性化に全く影響しなかった。しかしながら、偶然コントロールに使っていたNotch1-ICD(活性化型Notch1)による転写活性化をNLKが酵素活性依存的に抑制することがわかった。同様の現象は、ヒト大腸癌由来細胞SW480及びマウス神経芽細胞腫neuro-2aにおいても観察された。これらの結果から、NLKは細胞種を問わずNotch1-ICDによる転写活性化を特異的に抑制できることが示唆された。
続いて私たちは、「NLKによるNotch1シグナルの抑制」が「実際の個体でも起きているイベントなのか」を、モデル脊椎動物であるゼブラフィッシュを用いて検討した。Notchは脊椎動物の神経前駆細胞の維持に働いている。ゼブラフィッシュにおいては、体節形成期の神経板においてNotchシグナルが標的遺伝子her4及びhes5の発現を誘導し、これを介して神経前駆細胞が神経細胞になるのを抑制している。私たちはモルフォリノアンチセンスオリゴヌクレオチドを用い、ゼブラフィッシュにおいてNLKを機能阻害し、NotchシグナルとNLKの関係を検討した。NLK機能阻害胚(NLK k/d)では、her4及びhes5の発現が上昇し、神経細胞が減少した。また逆に、NLKを過剰発現すると、her4の発現が激減し、神経細胞が増加した。これらの結果から、「脊椎動物の神経発生過程において、NLKがNotchシグナルを抑制し、神経細胞形成を促進していること」が示唆された。
次に、「Notch1リン酸化がどのようなメカニズムでNotchシグナル低下を引き起こすのか?」を検討した。NLKによるNotch1のリン酸化は、Notch1-ICDの「タンパク質安定性制御」や「細胞内局在制御」には影響を与えなかった。そこで私たちは、NLKによるリン酸化は「Notch1-ICDのMAM、CSLとの三者複合体形成」に影響を与えているのではないかと考えた。neuro-2a細胞にMycタグ付きのNotch1-ICDを単独、あるいはNLKと共に発現させて抗Myc抗体で免疫沈降し、Notch1-ICDと細胞内在性のMAM、CSLの共沈降を調べた。その結果、NLKによるNotch1-ICDのリン酸化がNotch1-ICDとMAM、あるいはCSLとの結合を低下させることが明らかになった。さらに、ゲルシフトアッセイを行い、NLKによってリン酸化されたNotch1-ICDがDNA上でMAM、CSLとの三者複合体を形成できないことを確認した。
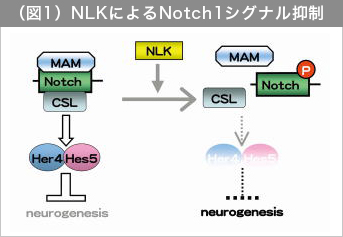 続いて私たちは、NLKによるNotch1のリン酸化部位を同定し、NLKのリン酸化を受けないNotch1変異体(リン酸化部位のセリン残基をアラニン残基に置換したA mutant)と擬似リン酸化状態のNotch1変異体(リン酸化部位のセリン残基をアスパラギン酸残基に置換したD mutant)を作製し、これらのNotchシグナル及び神経細胞形成への影響を調べた。Neuro-2a細胞とゼブラフィッシュの両方において、A mutantは野生型Notch1よりも強く標的遺伝子の発現を誘導し、神経細胞形成も強く抑制した。逆にD mutantは野生型Notch1よりも標的遺伝子発現誘導能が弱く、かつ神経細胞形成の抑制能も弱かった。
続いて私たちは、NLKによるNotch1のリン酸化部位を同定し、NLKのリン酸化を受けないNotch1変異体(リン酸化部位のセリン残基をアラニン残基に置換したA mutant)と擬似リン酸化状態のNotch1変異体(リン酸化部位のセリン残基をアスパラギン酸残基に置換したD mutant)を作製し、これらのNotchシグナル及び神経細胞形成への影響を調べた。Neuro-2a細胞とゼブラフィッシュの両方において、A mutantは野生型Notch1よりも強く標的遺伝子の発現を誘導し、神経細胞形成も強く抑制した。逆にD mutantは野生型Notch1よりも標的遺伝子発現誘導能が弱く、かつ神経細胞形成の抑制能も弱かった。
また、D mutantは三者複合体形成能力が低下していた。これらの結果から、NLKによるNotch1“リン酸化”の重要性を分子レベル(三者複合体形成の阻害)と個体レベル(神経細胞形成)をリンクさせて示すことが出来た(図1)。
本成果は、Nature Cell Biology誌2010年3月号ににおいて論文として発表した。
神経成長因子Nerve Growth Factor (NGF)は感覚神経等の生存や軸索伸展において重要な機能を果たす細胞外シグナル分子である。また、NGFはラット神経細胞様細胞株 PC12に神経突起を誘導する能力を持つことがよく知られている。PC12細胞は、NGFシグナルの細胞内情報伝達経路を解析するためのモデルシステムとしてよく使用されている。現在までに、NGFの下流でERKやp38といったMAPK群が活性化し、神経突起伸長に必須の働きをしていることが知られている。NLKはMAPKと類似の構造を持つ酵素であり、また、PC12細胞及び脊椎動物の神経組織に強く発現していることから、NLKもMAPK同様にNGFシグナルの下流で機能する可能性が期待できる。そこで、この可能性を検討した。
まず私たちは、PC12細胞にNGFを添加すると、その後3-60分の間、NLKの酵素活性が上昇し、かつNLKが細胞質から核内と細胞辺縁部へ局在変化することを見いだした。また、NGFが、低分子量GTPase Rasを介してNLKのリン酸化を引き起こすこともわかった。さらに、NGFと類似の活性を持つ成長因子である上皮成長因子EGFによっても、Ras依存的にNLKのリン酸化が引き起こされることがわかった。
つぎに、NGFシグナルによって活性化されたNLKがどのような分子をリン酸化しているのかを調べた。これまでに、NGF刺激直後にリン酸化を誘導されるタンパク質として、MAP1Bとパキシリンが知られている。NGFの下流でこれらをリン酸化するのはタンパク質リン酸化酵素GSK-3βであると考えられていた。何故ならば、PC12細胞をあらかじめGSK-3β阻害剤として知られる塩化リチウムで処理しておくと、NGFで刺激してもMAP1Bとパキシリンのリン酸化が起きなくなるからである。しかしながら一方で、GSK-3βはNGF刺激によって不活性化されることも報告されていた。このことから、「GSK-3βではない、NGF刺激直後に活性化する塩化リチウム感受性なリン酸化酵素」がNGF刺激によるMAP1Bおよびパキシリンのリン酸化誘導に関与することが予測される。私たちは、NLKこそがこの酵素ではないかと仮定し、その仮説を検証した。まず、NLKがNGFの下流でMAP1B及びパキシリンのリン酸化に関与しているかを検討した。PC12細胞においてNLKを siRNAを用いてノックダウンしたところ、NGF 刺激によるパキシリンのセリン126とMAP1Bのリン酸化が低下した。また、Co-IPアッセイによりNLKがパキシリン、MAP1Bの双方に直接結合することが判明し、さらにin vitro kinase assayによりNLKがパキシリン、MAP1Bの双方を直接リン酸化することが確認された。これらの結果から、NLKがNGFの下流で働いて、MAP1Bとパキシリンをリン酸化することが示唆された。加えて、PC12細胞に塩化リチウムを10-50mM添加するとNGF によるNLKの活性化が阻害されること、in vitroで塩化リチウムがNLKの活性を直接阻害することを見いだした。このように、私たちの仮説が正しいことが証明された。
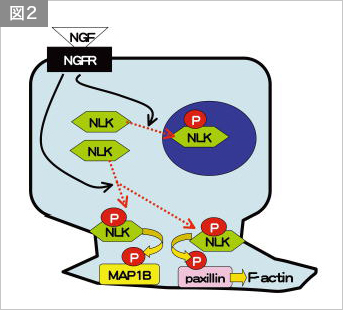 続いて、NLKによるパキシリンリン酸化の意義を検討した。パキシリンはfocal adhesionという細胞接着において、細胞外マトリクスとアクチン骨格をリンクさせる重要なタンパク質である。上述のように、NGFをPC12細胞に添加すると、その5-30分後にNLKは細胞辺縁部に局在する。私たちは、NLK同様にNGF添加5-30分後に細胞辺縁部にセリン126をリン酸化されたパキシリンとアクチン繊維が集積することを見いだした。また、NLKをRNAiすると、この「セリン126をリン酸化されたパキシリンとアクチン繊維の集積」が失われ、神経突起伸長が起きなくなることを見いだした。即ち、「NGFによって活性化されたNLKは細胞辺縁部に移動し、そこでパキシリンのセリン126のリン酸化とアクチン繊維の集積誘導を行い、神経突起伸長に貢献することが示された。
続いて、NLKによるパキシリンリン酸化の意義を検討した。パキシリンはfocal adhesionという細胞接着において、細胞外マトリクスとアクチン骨格をリンクさせる重要なタンパク質である。上述のように、NGFをPC12細胞に添加すると、その5-30分後にNLKは細胞辺縁部に局在する。私たちは、NLK同様にNGF添加5-30分後に細胞辺縁部にセリン126をリン酸化されたパキシリンとアクチン繊維が集積することを見いだした。また、NLKをRNAiすると、この「セリン126をリン酸化されたパキシリンとアクチン繊維の集積」が失われ、神経突起伸長が起きなくなることを見いだした。即ち、「NGFによって活性化されたNLKは細胞辺縁部に移動し、そこでパキシリンのセリン126のリン酸化とアクチン繊維の集積誘導を行い、神経突起伸長に貢献することが示された。
本成果は(図2)、Journal of Neurochemistry誌において論文として発表した。
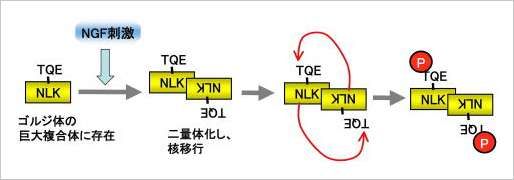
NLKは、STAT3やTcf/Lef、Notch1など重要な転写制御因子をリン酸化してその活性を制御する能力を持っている。 しかしながら、NLKの活性制御機構はほとんどわかっていない。私たちは、NLKのホモ二量体形成がNLKの活性化とNLKの核局在に必須であることを発見した。私たちはまず、生化学的解析により、NLKがホモダイマーを形成し、intermolecular mannerで自身のThr-286をリン酸化することを見いだした。さらに、この自己リン酸化がNLKの活性化に必須であることを発見した。また、線虫C. elegansにおけるNLKのホモログlit-1の機能欠損変異に相当する部位であるCys-425に変異を導入したNLKはホモダイマーを形成できないだけでなく、酵素活性も核への局在活性も失っていた。これに対して、既知のNLK活性化因子である神経成長因子NGFによって細胞を刺激すると、細胞内在性のNLKがダイマー形成し、Thr-286の自己リン酸化を行なうことがわかった。加えて、NLKのダイマー形成と自己リン酸化がNGF刺激によって誘導されるPC12細胞の神経突起伸長に必須であることがわかった。これらの結果から、NLKのダイマー形成がNLKの機能的活性化の「最初のカギ」となることが示唆された。
このように全く未解明であったNLKの活性制御メカニズムの中核部分を解明することに成功した。
背景1)Wnt/βカテニンシグナルの細胞内における情報伝達機構
Wnt/βカテニンシグナルは種を超えて保存されており、動物の組織構築/器官形成だけではなく、幹細胞性の維持や癌発生など様々な局面で重要な役割を担っている。その為、Wntシグナルの機能および制御機構の解明は、新たな治療法の開発や創薬につながる可能性があると期待されており、現在までに数多くの研究者がその解明に尽力してきた。その結果として、図1に示すリニアなシグナル伝達経路を軸に、複雑なネットワークの存在が示唆されている。
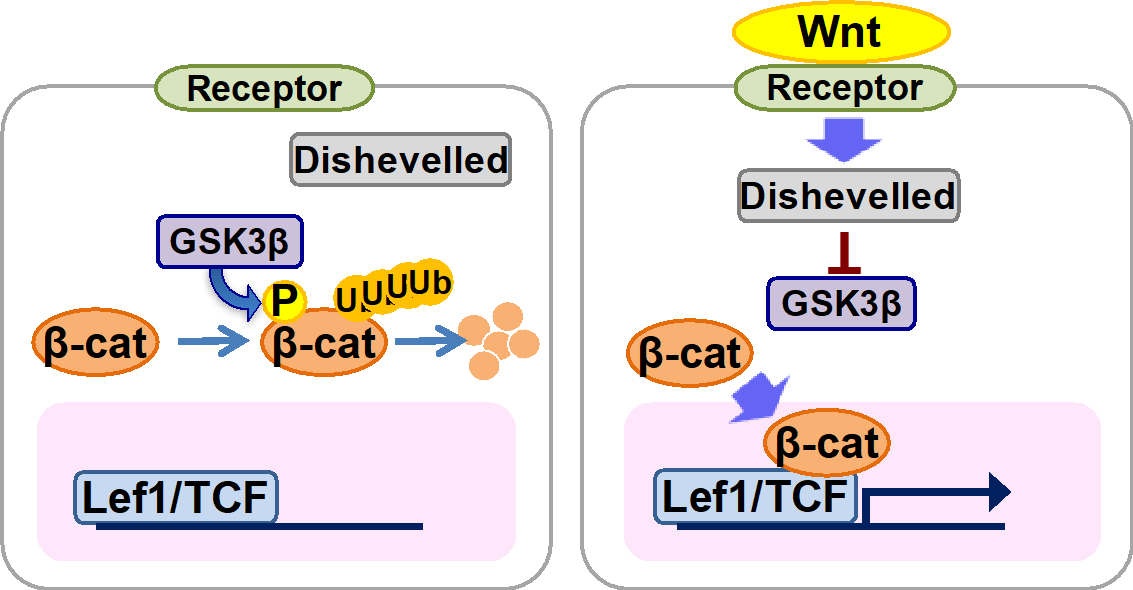
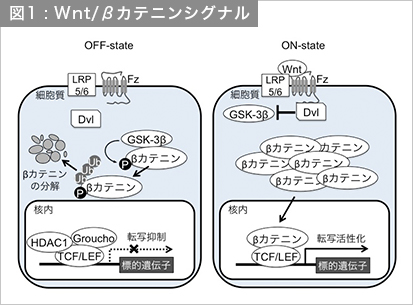 Wnt/βカテニンシグナルは、転写因子TCF/LEF (T-cell factor/Lymphoid enhancer factor)のコアクチベーターであるβカテニンの細胞質中におけるタンパク質量を調節することにより標的遺伝子の発現を制御している。Wntシグナルを受容していない細胞では、βカテニンは、そのN末端領域をタンパク質リン酸化酵素GSK-3β (glycogen synthase kinase-3β)によってリン酸化され、そのリン酸化の結果としてユビキチン化される。そして、ユビキチン化されたβカテニンはプロテアソームによって分解される。この時、転写因子であるTCF/LEFはGrouchoなどのコリプレッサーやヒストン脱アセチル化酵素HDAC1と複合体を形成し、Wnt/βカテニンシグナル標的遺伝子の転写を抑制している。細胞が細胞間情報伝達分子Wntを受容すると、そのシグナルはFzやLRP5/6からなる受容体複合体を介してDishevelled (Dvl) タンパク質に伝達され、続いてDvlはGSK-3βの活性を抑制する。その結果、βカテニンはGSK-3βによるリン酸化を免れて細胞質中に蓄積し、核内に移行してTCF/LEFと複合体を形成し、標的遺伝子の転写を活性化する(図1)。
Wnt/βカテニンシグナルは、転写因子TCF/LEF (T-cell factor/Lymphoid enhancer factor)のコアクチベーターであるβカテニンの細胞質中におけるタンパク質量を調節することにより標的遺伝子の発現を制御している。Wntシグナルを受容していない細胞では、βカテニンは、そのN末端領域をタンパク質リン酸化酵素GSK-3β (glycogen synthase kinase-3β)によってリン酸化され、そのリン酸化の結果としてユビキチン化される。そして、ユビキチン化されたβカテニンはプロテアソームによって分解される。この時、転写因子であるTCF/LEFはGrouchoなどのコリプレッサーやヒストン脱アセチル化酵素HDAC1と複合体を形成し、Wnt/βカテニンシグナル標的遺伝子の転写を抑制している。細胞が細胞間情報伝達分子Wntを受容すると、そのシグナルはFzやLRP5/6からなる受容体複合体を介してDishevelled (Dvl) タンパク質に伝達され、続いてDvlはGSK-3βの活性を抑制する。その結果、βカテニンはGSK-3βによるリン酸化を免れて細胞質中に蓄積し、核内に移行してTCF/LEFと複合体を形成し、標的遺伝子の転写を活性化する(図1)。
背景2)NLKによるTCF/LEFのリン酸化
Wnt/βカテニンシグナルは、線虫C. elagansやショウジョウバエなどの無脊椎動物から、カエルや魚、ヒトを含む脊椎動物まで高度に保存されている。私は、名古屋大学松本邦弘教授の研究室の大学院生であった時に、線虫を用いた遺伝学的解析(オレゴン大学Bruce Bowerman教授らとの共同研究)により、LIT-1と呼ばれるタンパク質リン酸化酵素がTCF/LEFを負に制御することを発見している(Meneghini, Ishitani et al., Nature,1999)。また私たちは、「ヒト胎児腎臓由来細胞株HEK293や子宮頸癌由来細胞株HeLaにおいても、哺乳動物LIT-1ホモログであるNLKがTCF/LEFの活性を負に制御すること」を発見した(Ishitani et al., Nature, 1999; Ishitani et al., MCB, 2003)。具体的には、NLKがin vitroにおいてTCF/LEFファミリーの転写因子であるLEF1のThr-155及びThr-166やTCF7L2のThr-178及びThr-189をリン酸化すること、NLKにリン酸化されたTCF7L2はリン酸化されていないTCF7L2に比べ、in vitroにおけるDNA結合能力が低いこと、活性型βカテニンの過剰発現によって誘導されるWnt/βカテニンシグナルレポーター“TOPFLASH”の活性化をNLKの過剰発現によって抑制することができることを示し、これらのことから「NLKがTCF/LEFをリン酸化することによりTCF/LEFのDNA結合能を低下させ、その結果としてTCF/LEF依存的な遺伝子発現が抑制される」というモデルを提唱した。しかしながら、その後十年近く、TCF/LEFリン酸化の脊椎動物における生理学的意義は全く不明のままであった。
結果1)NLKはゼブラフィッシュ中脳においてLEF1のリン酸化を介してWnt/βカテニンシグナルを正に制御し、神経前駆細胞の増殖を促す。
小型魚類ゼブラフィッシュは近年in vivoイメージングに適したモデル動物として注目されており、これまでに、TCF/LEFの活性依存的に不安定化GFP(dGFP)を発現するレポーター遺伝子が組み込まれたゼブラフィッシュ系統TOPdGFP-fishが作成されている。私は、NLKによるTCF/LEFリン酸化の脊椎動物における意義を探るために、まずこのTOPdGFP-fishを用いて「NLKがTCF/LEFの活性を制御する組織、細胞」の同定を試みた。TOPdGFP-fishは、受精後18−20時間ごろから中脳領域において強くdGFPを発現するが、モルフォリノアンチセンスオリゴ(MO)を用いてゼブラフィッシュNLKホモログNlk2の機能を阻害したところ、驚くべきことに、受精後24時間以降の中脳領域のdGFP発現が低下した。以前のHEK293、HeL細胞の解析から「Nlk2がTCF/LEFの活性を負に制御すること」が予測されていたが、このゼブラフィッシュを用いた実験の結果は、想定外なことに、「Nlk2が受精後24時間以降の中脳領域においてTCF/LEFの活性を正に制御すること」を示唆している。続いて、Nlk2によるTCF/LEF活性促進の意義を探るために、Nlk2機能阻害ゼブラフィッシュの中脳を継続的に観察したところ、Nlk2機能阻害胚では、中脳領域の神経前駆細胞の増殖が減少し、結果として、最終的に形成される中脳視蓋のサイズが小さくなってしまうことが分かった。また、中脳領域で発現するTCF/LEFファミリーの転写因子であるLEF1をMOで機能阻害すると、Nlk2機能阻害と同様に中脳領域においてdGFPの発現低下と、神経前駆細胞の増殖低下、中脳視蓋のサイズ縮小が観察された。これらの結果から、「LEF1活性のNlk2による正の制御が中脳における神経前駆細胞の増殖と適切な大きさの視蓋の形成に必須である」と考えられる。
次に私たちは、Nlk2がLEF1のリン酸化を介してLEF1活性を促進しているかどうかを検討した。まず、培養細胞株においてNlk2がゼブラフィッシュLEF1をリン酸化することを確認した。次に、リン酸化されたLEF1を特異的に認識する抗体を作成し、これを用いてゼブラフィッシュ胚における内在性LEF1のリン酸化の検出を試みたところ、Nlk2依存的なLEF1の活性化が観察される受精後24時間胚において内在性LEF1のリン酸化を検出することができた。また、このリン酸化は、Nlk2機能阻害胚では検出されなかったことから、受精後24時間胚においてNlk2がLEF1をリン酸化していると考えられる。続いて、レスキュー実験を行い、ゼブラフィッシュ胚中脳においてNlk2がLEF1のリン酸化を介して機能しているどうか調べた。Nlk2機能阻害胚の中脳視蓋サイズ縮小の表現型は、野生型Nlk2や疑似リン酸化LEF1変異体(リン酸化部位のThr残基をグルタミン酸に置換した変異体)の強制発現によりレスキューされたが、酵素活性を欠くNLK変異体や野生型LEF1を強制発現してもレスキューされなかった。これらの結果は、Nlk2がLEF1のリン酸化を介してゼブラフィッシュ中脳視蓋の正常な形成に貢献することを示唆している。以上の結果をまとめると、ゼブラフィッシュ中脳においてNLKはLEF1のリン酸化を介してWnt/βカテニンシグナル(LEF1活性)を正に制御し、神経前駆細胞の増殖を促し、適切なサイズの中脳視蓋形成に貢献していると考えられる。
結果2)NLKは神経前駆細胞において、LEF1のリン酸化を介してHDAC1によるLEF1活性抑制を解除し、LEF1の転写活性を促進する。
上述のように、NLKはHeLa細胞及びHEK293細胞においてはTCF/LEFの活性を抑制する。しかしながら今回私たちは、ゼブラフィッシュ中脳においてNLKがLEF1を正に制御することを見いだした。では、このようなNLKによるLEF1の正の制御は、哺乳動物の神経組織の細胞においても起きるのだろうか?私たちは、この可能性を神経前駆細胞様の細胞株である、マウスneuro-2a細胞とラットPC12細胞を用いて解析した。その結果、これらの細胞では、活性型βカテニン、あるいは活性型βカテニンとLEF1を過剰発現しても、Wnt/βカテニンシグナルレポーター“TOPFLASH”の活性化やWnt/βカテニンシグナル標的遺伝子CyclinD1の発現誘導が起きないこと、活性型βカテニンとLEF1とともにNLKを過剰発現すると、TOPFLASHの活性化やCyclinD1の発現誘導が起こることが分かった。また、リン酸化部位(Thr-155及びSer-166)をアラニンに置換したLEF1-2A変異体はNLK及び活性型βカテニンとともに発現させてもTOPFLASHを活性化できず、一方でThr-155とSer-166をグルタミン酸に置換して疑似リン酸化状態にしたLEF1-2E変異体を活性型βカテニンとともに発現させると、NLK非存在下でもTOPFLASHを活性化した。従ってこれらの結果から、NLKはLEF1リン酸化を介してβカテニン-LEF1複合体の転写活性を活性化すると考えられる。
次に、NLKによるLEF1リン酸化がどのような分子機構でLEF1を活性化するのかを探った。興味深いことに、NLKがLEF1の活性を負に制御する細胞であるHeLa細胞やHEK293細胞では、活性型βカテニンとLEF1を過剰発現しただけでTOPFLASHの活性化が起こる。このことから、「神経前駆細胞様細胞株ではHeLa細胞やHEK293細胞には存在しないLEF1抑制機構が働いており、NLKはこの抑制機構を阻害することによりLEF1の活性を促進するのではないか」と推測した。これまでに、マウスの脳やゼブラフィッシュの網膜等、脊椎動物の神経組織においてヒストン脱アセチル化酵素HDAC1がTCF/LEFに結合してTCF/LEFの転写活性を負に制御するという報告があった。そこで、LEF1抑制機構の実体がHDAC1である可能性を検討した。活性型βカテニンとLEF1のみを過剰発現したneuro-2a細胞をHDAC1阻害剤であるトリコスタチンA(TSA)で処理したところ、TOPFLASHの活性が強く上昇した。一方、活性型βカテニンとLEF1を過剰発現したHEK293細胞やHeLa細胞にTSAを添加しても、TOPFLASH活性はそれ以上に上昇しなかった。従って、神経前駆細胞様細胞株特異的に存在するLEF1抑制機構の実体はHDAC1であると考えられた。続いて、NLKがHDAC1によるLEF1阻害を解除する可能性を検討するために、NLK活性とHDAC1-LEF1複合体形成能の関係をCo-IPにより調べた。その結果、NLKを過剰発現したneuro-2a細胞ではLEF1とHDAC1の結合が低下すること、リン酸化を受けないLEF1-2A変異体はNLK存在下でもHDAC1と強く結合すること、擬似リン酸化変異体LEF1-2EはNLK非存在下でもHDAC1とほとんど結合できないことが明らかになった。これらの結果から、NLKはLEF1をリン酸化することにより、HDAC1とLEF1の結合を低下させ、LEF1の転写活性を上昇させると考えられる。また、ゼブラフィッシュNlk2阻害胚の中脳におけるTOPdGFPレポーター活性の低下は、TSA処理あるいはMOによるHDAC1機能阻害により回復した。したがって、NLKはゼブラフィッシュ中脳においてもHDAC1によるLEF1抑制を解除することでLEF1活性を促進していると考えられる。
結果3)神経前駆細胞においては、Wnt-1/Wnt-3aはDvlを介してNLKを活性化する。
私たちは最後に、NLKによるLEF1のリン酸化がどのような上流シグナルによって活性化されるかを検討した。受精後24時間のゼブラフィッシュ中脳においてWnt1が強く発現していたことから、「Wnt1ファミリーの細胞外リガンドがNLKの上流シグナルである」という仮説を立て、その仮説の検証を行った。Wnt1をMOにより機能阻害したゼブラフィッシュ胚では、Nlk2機能阻害胚と同様に、受精後24時間における中脳のTOPdGFP活性とLEF1リン酸化レベルが低下し、また、中脳に形成される視蓋のサイズが縮小することが分かった。加えて、PC12細胞にWnt1ファミリーの分子であるWnt-3aのシグナルを入力すると、NLKの酵素活性の活性化、LEF1リン酸化、LEF1とHDAC1の結合の低下、TOPFLASHレポーターの活性化が起きること、Wnt-3aシグナルによるLEF1リン酸化とTOPFLASHの活性化がNLKのRNAiによって阻害されることを見いだした。これらの結果から、NLKによるLEF1リン酸化は、Wnt-1/Wnt-3aシグナルの下流で起こると考えられる。さらに私たちは、PC12細胞ではWnt-3aシグナル依存的にWnt/βカテニンシグナルのメディエーターであるDvlとNLKの結合が増加すること、neuro-2a細胞やPC12細胞にDvl1を過剰発現するとLEF1リン酸化とTOPFLASHの活性化が起き、NLKをRNAiするとこれらが起きなくなることを発見した。従って、神経前駆細胞においてWnt-1/Wnt-3aシグナルはDvlを介してNLKを活性化すると考えられる。また、ここまでの結果をあわせて考えると、「神経前駆細胞においては、WntシグナルはDvlの下流でβカテニンの安定化とNLKの活性化の双方を導き、それによりLEF1を活性化する」と考えられる。
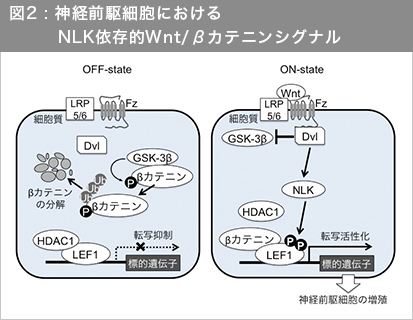 以上の結果から図2に示すモデルを提唱する。
以上の結果から図2に示すモデルを提唱する。
「Wnt分子を受容していない神経前駆細胞においては、HDAC1がLEF1に結合しており、そのため、LEF1の転写活性は低く抑えられている。Wnt分子を受容した神経前駆細胞では、Dvlが、GSK-3βの働きを抑えて核内のβカテニン-LEF1複合体の形成を誘導する一方で、NLKと結合してNLKを活性化する。活性化したNLKはLEF1をリン酸化してLEF1とHDAC1の結合を低下させ、βカテニン-LEF1複合体の転写活性を促進し、これにより神経前駆細胞の増殖を促し、適切なサイズの神経組織の構築に貢献する」
本研究の結果は、「同様の翻訳後修飾のアウトプットが細胞種によって異なること」を示している。このことは、シグナル伝達における“cell context”の重要性、即ち、「自分が解析している翻訳後修飾の機能と制御が“普遍的”なものであるか、あるいは“組織・細胞特異的”なものであるか、慎重に考えるべきである」ということを改めて思い知らせてくれる。今後もこの点に留意し、細胞株とモデル動物の双方を用いて研究を進めていきたい。
上述の通りWntシグナルは多様かつ重要な機能を担うシグナル経路であるが、この経路が個体のいつ、どこで、どの程度の強さで働くかは十分に分かっていない。そこで、Wntシグナル活性の活動を高感度に検出できるレポーターを作成し、これをモデル動物ゼブラフィッシュに組み込み、Wntシグナルの活動の時空間的動態を明らかにした(このホームページのトップページに例を示した)。
さらにこのレポーターゼブラフィッシュを用い、Wntシグナルが魚類のエラの器官である鰓弁の伸長や、頭部や背面の側線(水流を感知する器官)の形成に必須であることを見出した。これらの器官は魚類や両生類にしか存在しないが、本成果は、人が持つ「伸長を伴って形成される器官(生殖器など)」や「感覚器官」の形成機構を理解する一助となる可能性もある。
また、このレポーターゼブラフィッシュにより、肝臓や膵臓、視床下部などといった体の深部に存在する臓器の形成過程や、器官の再生・維持過程におけるWntシグナルの活動の観察が可能になった。今後このレポーターシステムを用いることで、臓器・器官の構築・維持・再生過程の分子レベルでの理解が促進されることが大いに期待できる。加えて、我々が作成したレポーターは動物種を超えて使用できることを確認しており、哺乳動物や無脊椎動物など他の動物におけるWntシグナル研究、器官構築研究にも貢献しうると期待している。
研究の背景
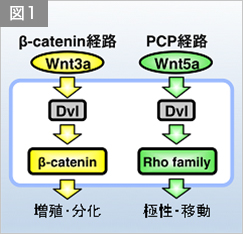 細胞外情報伝達分子Wntは、動物の個体発生と恒常性の維持において、細胞の増殖・分化・極性・移動を制御します。Wntの情報伝達の破綻は、種々の先天性の疾患やがんなどの成人病の発症の原因となります。哺乳類では19種類のWnt分子が同定されており、Wnt3aファミリーとWnt5aファミリーの二つに大別されます。細胞はWntMacintosh HD:Users:tohruishitani:Desktop:医科歯科図.pdf3aファミリーの分子を受容すると、その情報を細胞内情報伝達分子Dishevelled(Dvl)タンパク質へと伝えます(図1左)。これにより活性化したDvlはβ-catenin経路を活性化します。β-catenin経路が活性化すると、細胞質中でβ-cateninタンパク質の安定化が起き、結果として細胞の増殖や分化が誘導されます。一方、細胞がWnt5aファミリーの分子を受容した場合も、Dvlが活性化しますが、Wnt5aの下流で活性化したDvlはPCP経路を活性化します。PCP経路が活性化すると、Rhoファミリーの低分子量Gタンパク質群の活性調節が起き、結果として細胞の極性や移動が制御されます(図1右)。このように、Dvlは、Wntシグナルをβ-catenin経路とPCP経路に振り分ける、Wntシグナルの“HUB”分子です。興味深いことに、様々な腫瘍組織においてDvlタンパク質の増加とそれに伴うWntシグナルの異常活性化が観察されており、このことから、Dvlのタンパク質安定性の厳密な制御がWntシグナルの制御と組織恒常性維持に必須であると推測されています。しかしながら、Dvlの安定性制御機構は未だによくわかっていません。
細胞外情報伝達分子Wntは、動物の個体発生と恒常性の維持において、細胞の増殖・分化・極性・移動を制御します。Wntの情報伝達の破綻は、種々の先天性の疾患やがんなどの成人病の発症の原因となります。哺乳類では19種類のWnt分子が同定されており、Wnt3aファミリーとWnt5aファミリーの二つに大別されます。細胞はWntMacintosh HD:Users:tohruishitani:Desktop:医科歯科図.pdf3aファミリーの分子を受容すると、その情報を細胞内情報伝達分子Dishevelled(Dvl)タンパク質へと伝えます(図1左)。これにより活性化したDvlはβ-catenin経路を活性化します。β-catenin経路が活性化すると、細胞質中でβ-cateninタンパク質の安定化が起き、結果として細胞の増殖や分化が誘導されます。一方、細胞がWnt5aファミリーの分子を受容した場合も、Dvlが活性化しますが、Wnt5aの下流で活性化したDvlはPCP経路を活性化します。PCP経路が活性化すると、Rhoファミリーの低分子量Gタンパク質群の活性調節が起き、結果として細胞の極性や移動が制御されます(図1右)。このように、Dvlは、Wntシグナルをβ-catenin経路とPCP経路に振り分ける、Wntシグナルの“HUB”分子です。興味深いことに、様々な腫瘍組織においてDvlタンパク質の増加とそれに伴うWntシグナルの異常活性化が観察されており、このことから、Dvlのタンパク質安定性の厳密な制御がWntシグナルの制御と組織恒常性維持に必須であると推測されています。しかしながら、Dvlの安定性制御機構は未だによくわかっていません。
一方、私たちは以前に、ヒト培養細胞株HeLaを用いた解析により、β-catenin経路の正の制御因子としてタンパク質リン酸化酵素Hipk2を見つけていました。しかしながら、Hipk2によるβ-catenin経路促進機構の詳細やこの制御の生理学的意義は不明でした。
研究の成果
私たちはまず、モデル脊椎動物ゼブラフィッシュを用いて、Hipk2の生理機能を解析しました。Hipk2遺伝子の機能を阻害したゼブラフィッシュ胚では、胚発生過程を通じてWnt/β-catenin経路の標的遺伝子の発現の低下や、β-catenin経路によって制御されるシュペーマンオーガナイザーの形成や脳の後方化などの現象が全て異常になっていました。また意外なことに、Hipk2機能阻害胚では、Wnt/PCP経路によって制御される原腸胚形態形成運動にも異常が観察されました。これらの結果は、Hipk2が脊椎動物初期胚発生過程におけるβ-catenin経路とPCP経路の双方の活性化に必要であることを示しています。詳細な解析の結果、Hipk2機能阻害胚ではβ-catenin経路とPCP経路の共通の制御因子であるDvlタンパク質が劇的に減少していることが明らかになりました。つまり、Hipk2機能阻害胚では、Dvlタンパク質が失われているために、β-catenin経路とPCP経路双方の情報伝達に異常が生じたと推測されます(図2右)。
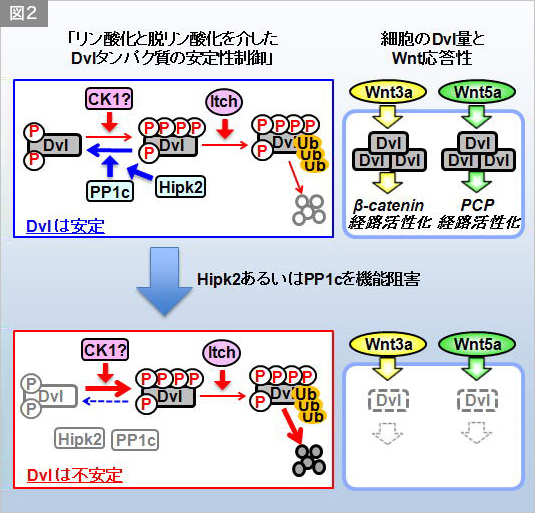
続いて、ヒト培養細胞株HeLaを用いてHipk2とDvlの関係を解析しました。その結果、HeLa細胞においてもHipk2をRNAiするとDvlタンパク質が減少すること、すなわち、ヒト細胞においても、Hipk2がDvlタンパク質の安定性に必須であることを見いだしました。
次に、Hipk2によるDvlの制御機構を生化学的手法を交えて解析しました。その結果、「Hipk2がDvlに結合し、Hipkがその酵素活性非依存的にタンパク質脱リン酸化酵素PP1cをDvlへとリクルートすること」と、「リクルートされたPP1cがDvlのC末端領域のCasein Kinase 1(CK1)リン酸化部位を脱リン酸化すること」、「この脱リン酸化が、ユビキチンリガーゼItchによるDvlのユビキチン化を阻害し、Dvlを安定化へと導くこと」を見いだしました(図2左)。この仮説と合致するように、PP1cを機能阻害したゼブラフィッシュやHeLa細胞では、Dvlタンパク質の減少が起き、β-catenin経路やPCP経路の伝達が低下しました(図2右)。また、Hipk2機能阻害によるDvlの減少とβ-catenin経路の活性低下は、Itchを同時に阻害することで回復しました。
最後に、Hipk2によるDvlの脱リン酸化部位を同定し、その部位をアラニン残基に置換したDvl 3A変異体(脱リン酸化状態のDvlの状態をミミックする変異体)を作製し、これを用いて、Hipk2がDvlの脱リン酸化を介してWntシグナルを制御しているのかを検討しました。Hipk2をRNAiしたHeLa細胞やHipk2を機能阻害したゼブラフィッシュ胚に野生型のDvlを発現させても、これが不安定なために、Hipk2阻害により生じるβ-catenin経路の活性低下を回復させることは出来ませんでしたが、Dvl 3A変異体を発現させた場合は、β-catenin経路の活性の回復が起きました。このことは、Hipk2がDvlの脱リン酸化と安定化を介してWntシグナルを正に制御することを示唆します。
このように、Wnt分子の細胞内情報伝達を支える、新たな情報伝達機構が明らかになりました。本発見により、Dvlタンパク質の安定性に作用するがん治療薬の開発など、新たながん治療法への道が開けると期待できます。
研究のキーワード
生命現象を理解するためのアプローチとして、最も説得力がある方法は「現象そのものを見ること」です。
近年、細胞の動きや分裂増殖、細胞間・細胞内で活動するシグナルなどのミクロの生命現象を蛍光や発光に変換して可視化する技術が開発され、これを用いて生きた動物の体内で起きている生命現象を目で見る(インビボイメージング)試みがなされています。
しかし、多くの生命科学研究で使用されているマウスやラットなどは体が大きく、また、透明度が低いため、これらを用いたインビボイメージングは容易ではありません。
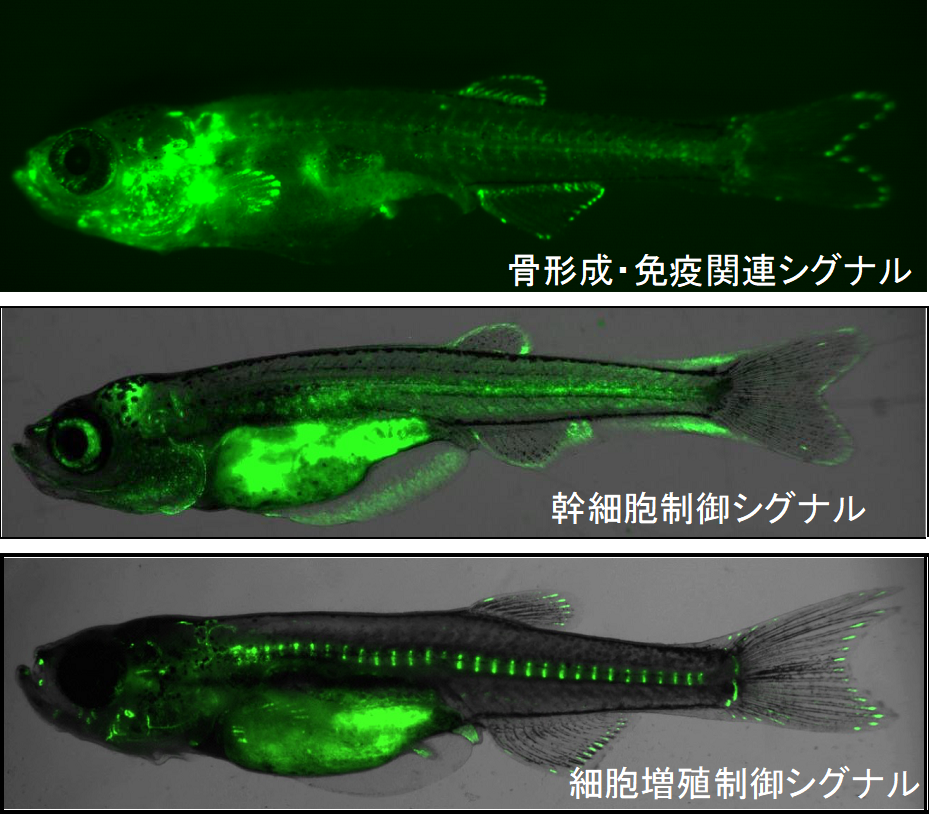
そこで、私たちの研究室では、体が小さく透明度が高い小型魚類を用いてインビボイメージングを行っています。特に細胞間・細胞内シグナル(写真)や癌細胞動態のイメージングを得意としています。
この技術を駆使して、マウスやラットを使った研究では到達し得ない「深さ」「精度」での生命現象の理解を目指しています。
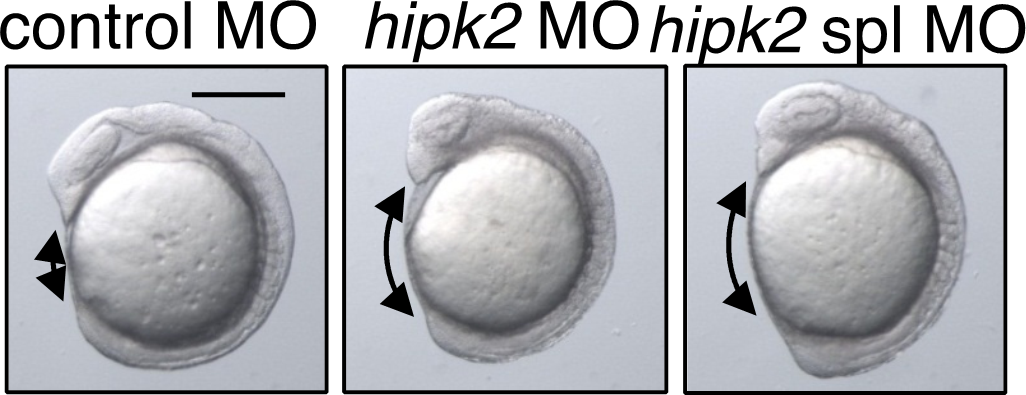
初期発生は、受精卵というたった一つの細胞から、赤ちゃんの体の基礎(胚)が生じるプロセスです。
このプロセスにおいては、細胞の増殖・分化・移動・死や組織のパターン形成や形態形成運動など多種多様な生命現象が同時多発的に起きます。
初期発生ののちに臓器の構築や維持、再生などといった形態形成現象が起きますが、これらの現象の大部分は初期発生で用いられたシステムを使いまわして実行されます。
即ち、初期発生を制御するシステムの理解は、動物の形態形成全般の理解に繋がると考えることができます。
私たちはこのような理由から、ゼブラフィッシュ初期発生をモデルとして、ヒトを含む動物のからだ作りを支えるシステムの探索と解析を行っています。
 動物や植物の発生(受精卵から機能する成体が構築されるプロセス)は、遺伝子情報やそれにプログラムされた分子の活動、化学反応の連鎖によって達成される。
動物や植物の発生(受精卵から機能する成体が構築されるプロセス)は、遺伝子情報やそれにプログラムされた分子の活動、化学反応の連鎖によって達成される。
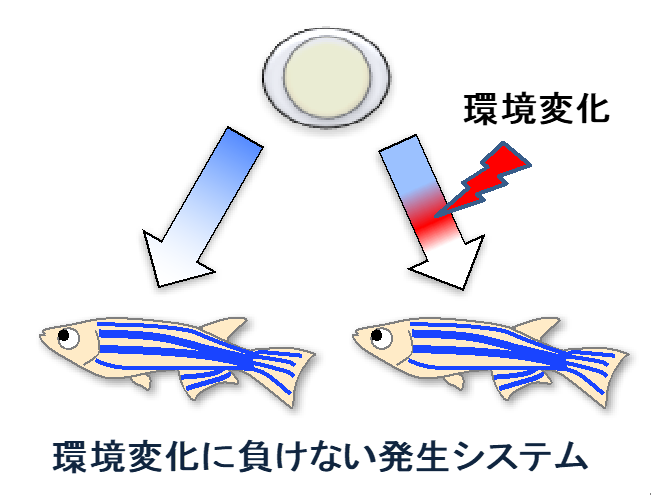
しかし、自然界の動植物は、その発生過程において様々な環境ストレス(急激な温度変化や有害な光線・物質など)にさらされていますが、それにも関わらず、見た目ほとんど違いのない形のからだを再現良く作り上げます。
このことは、動植物の発生システムには頑強性(ロバストネス)があることを示しています。
しかし、このロバストネスの分子実態はよくわかっていません。
私たちは、Wntシグナルによる組織パターン形成をモデルに、発生ロバストネスの実態解明を進めています。
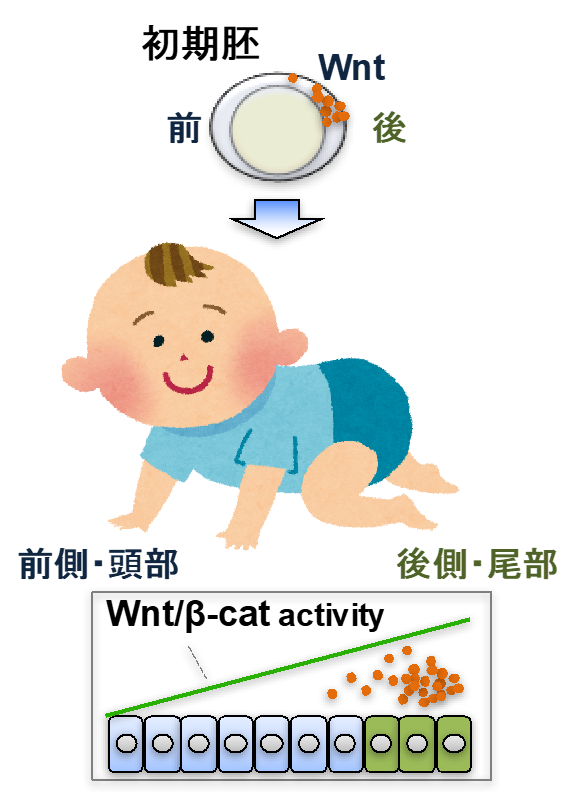 モルフォジェンシステムは、シグナル分子の濃度勾配依存的に組織内の細胞に位置情報を与える仕組みであり、動植物の組織パターン形成において必須の役割を果たします。
モルフォジェンシステムは、シグナル分子の濃度勾配依存的に組織内の細胞に位置情報を与える仕組みであり、動植物の組織パターン形成において必須の役割を果たします。
モルフォジェン研究の歴史は古く、1950年代から70年代にかけてチューリング、ウォルパート、クリックらによって概念的な土台が築かれ、その後の分子遺伝学の発展により、分子実体の大要が明らかにされました。
しかしながら、生きた組織内におけるモルフォジェンの拡散様式や濃度勾配形成の制御機構は未だによくわかっていません。
私たちは、モルフォジェンの一つであるWntに注目し、モルフォジェンの謎を解き明かそうとしています。
当研究室で現在精力的に研究を進めている、モルフォジェンシステムの破綻を防ぎ、正確な組織パターン形成を支える未知のシグナルシステム。
がんの克服は人類共通の夢であり、この夢に向けて、多くの研究者ががんの発症機序の解明とそれを基盤とした治療法開発に取り組んでいます。
特に近年、マウスや患者サンプルを使った解析により、がん発生に関わる遺伝子群・分子群や、がんの悪性化のメカニズムが急速に明らかになりつつあります。
しかしながら、これら従来のがん研究は“検出可能なレベルまで成長したがん”を対象としたものであり、“現行技術で検出が難しい発生初期のがん”についての理解はほとんど進んでいません。
私たちは、ゼブラフィッシュのインビボイメージング解析により、「体内に生じた“”単一の異常細胞(変異細胞)“からがんが生じる全プロセス」を明らかにしようとしています。
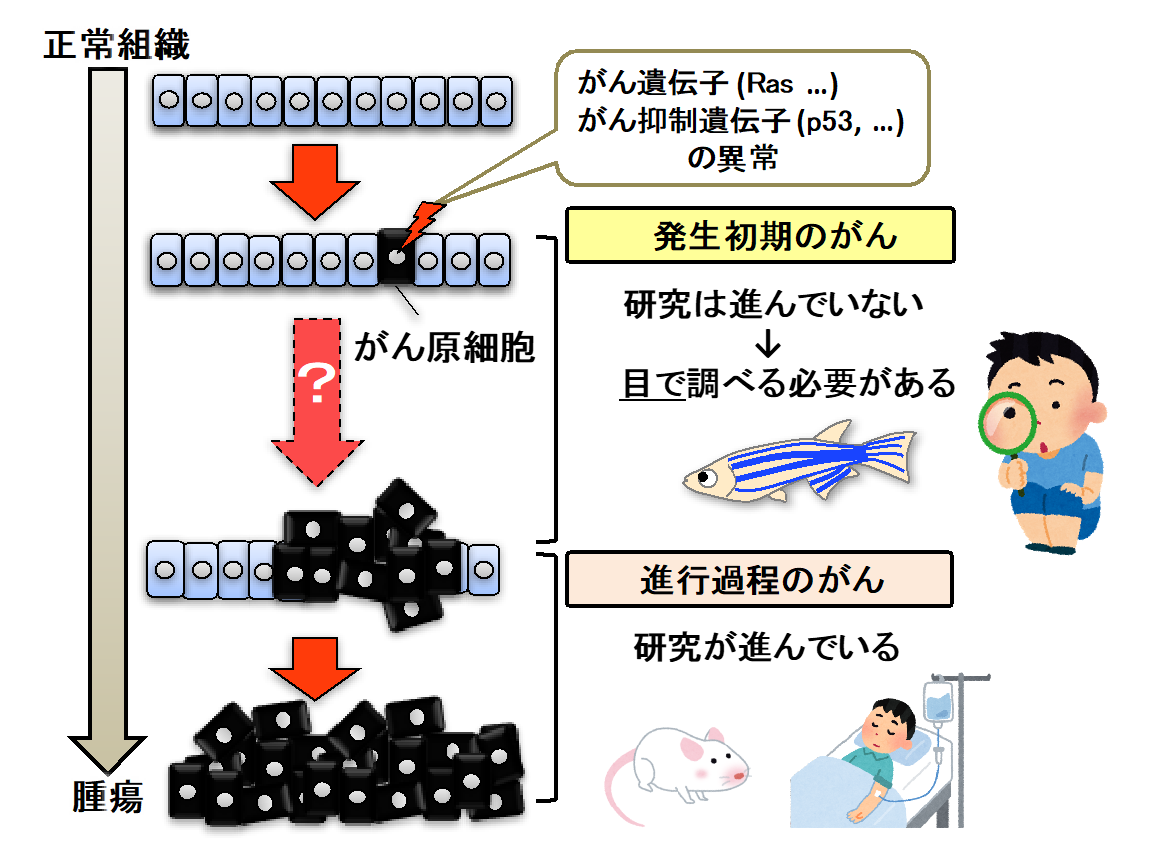
動物組織のなかで生じる、質的に異なる同種の細胞間の争い。
近年の研究により、組織に突発的に生じたがん細胞と隣接正常細胞が細胞競合を起こし、これが組織のがんに対する抵抗力やがんの進展の双方に関わることが明らかになりつつあります。
詳細は以下URLをご覧ください。
細胞外情報伝達分子Wntによって活性化される細胞シグナル。
Wntはいくつかのサブファミリーに分けられ、サブファミリーの違いや受容体の違いによって、異なる細胞内シグナル経路(Wnt/β-cateninシグナル、Wnt/PCP経路、Wnt/ Ca2+経路、Wnt/STOP経路など)を活性化します。
当研究室は特にWnt/β-cateninシグナル(図)に注目しています。
Wnt/β-cateninシグナルは、動物個体の発生及び恒常性維持の過程で繰り返し活性化し、状況に応じて細胞の増殖あるいは分化を促します。
また、組織幹細胞の維持においても必須の役割を果たします。
また、その多機能さゆえに、このシグナルの制御破綻は、がんや糖尿病、肥満、骨粗鬆症、精神疾患など多様な疾患の発症に関わります。
私たちの研究室では、Wnt/β-cateninシグナルの未知の制御機構を探索することで組織形態形成のメカニズム解明に貢献するするとともに、その制御技術を開発して医療に貢献することも目指しています。
Nemo-like kinase (NLK)は動物種をこえて保存されたMAPKファミリーのSer/Thrキナーゼです。
MAPKファミリーには、NLKの他にERK、p38、JNKがあり、この三種の解析は進んでいますが、NLKの研究は比較的遅れています。
私たちは、1999年に線虫においてWntシグナルを制御する酵素としてNLKを発見し、以来20年近くNLKの研究を行って来ました。
現在までに、私たち及び海外のグループなどの精力的な研究により、NLKが多様な分子をリン酸化して制御し、神経発生や神経変性疾患、がんの発症に関わることがわかって来ています。
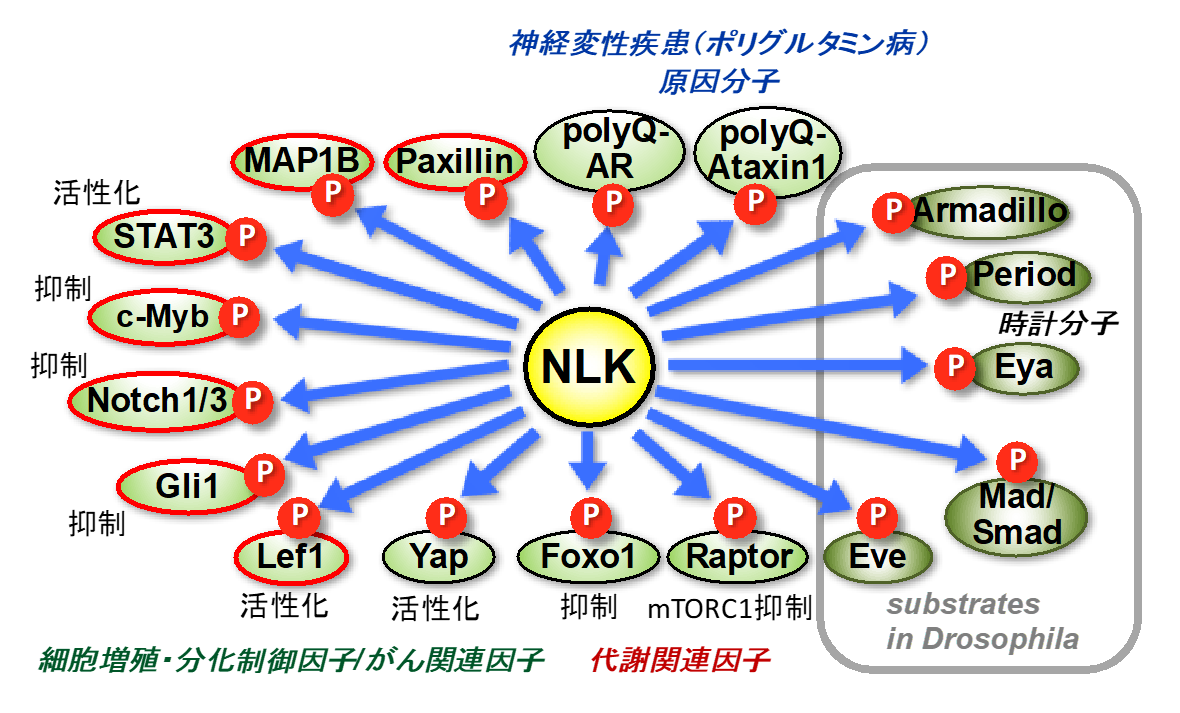
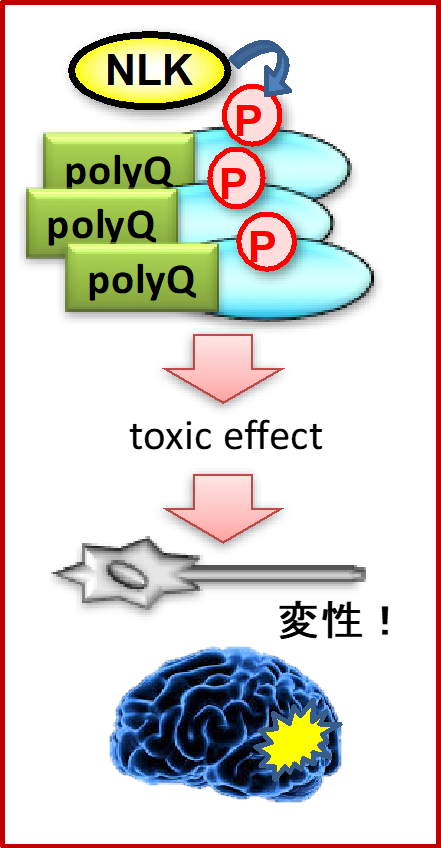 私たちの研究室では、NLKを起点に神経疾患の研究を進めています。
私たちの研究室では、NLKを起点に神経疾患の研究を進めています。
NLKは中枢神経組織に強く発現するキナーゼで神経発生において重要な役割を果たしますが、近年の研究により、NLKが神経疾患の発症に関わることを示唆する報告が相次いでなされています。
例えば、神経変性疾患の一種である脊髄小脳変性症一型や球脊髄性筋萎縮症のモデルマウスにおいてNLKの機能を阻害すると、神経変性の症状が顕著に回復されることが報告されています。
私たちは、これらの疾患におけるNLKの機能の詳細を明らかにするとともに、NLK阻害を介した新規神経疾患治療法を創り出したいと考えています。
病気のメカニズム解明や治療薬の開発を行うためには、ヒトの病気を再現した実験動物(疾患モデル)を使用する必要があります。
これまで、マウスやラットなどを疾患モデルとして用いた研究が精力的に行われ、近年ではマーモセットなどが注目されています。
これらの動物はヒトと同じ哺乳類であるという利点がある一方で、解析にあたってのスループットや検体数に制限があり、また、生命倫理的な問題によっても研究の制限があります。
私たちは、ゼブラフィッシュなどの小型魚類を疾患モデルとして使った、多検体、ハイスループット、高解像度(イメージングとの併用)で、かつ、生命倫理問題を軽減した疾患研究を提案しています。
治療薬の開発にあたっては、動物を使って薬の効果や毒性を評価する必要があります。
しかし、哺乳類モデルを使った評価をするには、薬を大容量準備する必要があり(高コスト)、かつ、多数の検体について行うのが困難です。
私たちの研究室では、ゼブラフィッシュなどの小型魚類を使った薬の評価を行っています。
小型魚類は体が小さいために使用する薬の量も少量(低コスト)ですみ、また水棲動物であるがゆえに「飼育シャーレに化合物を溶かすだけで薬が体内に取り込まれる」ため、薬の投与が容易です。
また、多産な動物ゆえに(ゼブラフィッシュであれば、数千個の検体を一度に準備可能)、多検体の評価が可能であり、生命倫理的な問題も比較的容易にクリアできます。現在までに海外のグループとのゼブラフィッシュ共同研究により、肝がんや白血病の治療薬候補が見つかって来ています。
私たちの研究は、初期発生やシグナル制御機構に注目し、魚をモデル動物として用いているがゆえに、医学薬学とは程遠い研究とみなされることもあります。
事実、PIの石谷は理学部出身で、度肝を抜くようなオモロイ発見をしたい、という強い思いを持っています。
一方で、私たちは自分たちが注目する生命現象はヒトの病気のメカニズム解明にも繋がるという確信を持って研究を進めており、かつ、自分たちが発見した現象や開発した実験システムを起点に疾患治療薬の開発に応用していこうとしています。
